Services
Jubilant Biosys’ Fragment-Based Drug Discovery Services
Fragment-based drug discovery refers to the discovery efforts by employing fragments as the starting points for a target of interest. Diverse fragments are experimentally determined to bind to the target of interest and are either expanded or grown or linked together to form potent larger compounds. Jubilant Biosys employs experiments based on Surface Plasmon Resonance (SPR) to determine fragments that bind to the target of interest. Complexes of the target and the fragments are experimentally generated either by X-Ray crystallography or Nuclear Magnetic Resonance (NMR) experiments or computationally determined by docking the fragments into protein structures or homology models.
Fragment Based Drug Discovery (FBDD):
FBDD – In this technique prior knowledge of 3 dimensional structure of protein target is a must. This is an unbiased approach, involving the construction of potent small molecule hits from low molecular mass fragments. There are two primary advantages as reported in Biochemistry (2012) 51, 4990-5003 by Scott D E et al: First, it is estimated that there are 1060−200 possible drug-like compounds (MW 300 – 500 Da) but only 107 possible molecules composed of up to 11 atoms of C, N, O and F that follow the rule-of-3. Thus, significantly larger portion of chemical space can be sampled with a fragment library (usually ~103 fragments) than ~105 – 106 larger molecules typical of a HTS campaign. Second, although fragment hits are weak-binding they must make high-quality interactions with the target to bind with sufficient binding affinity for detection. Because of these optimal interactions, fragments are very “atom-efficient” binders, demonstrated by high ligand efficiency (LE) i.e. –ΔG (Kcal/mole)/HA (non-hydrogen atoms).
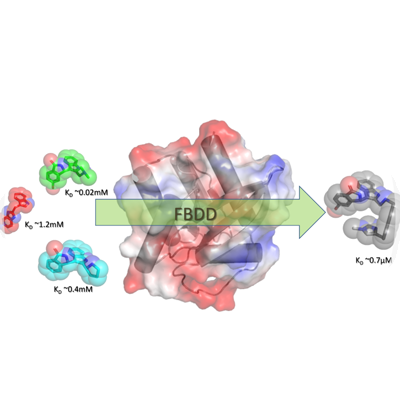
This technique involves generating a diverse fragment library from commercial sources for a given protein target by applying filters (i.e. natural product-likeness, fraction of SP3 centers, atom-pair fingerprints-to filter clusters) followed by affinity screening of fragments against target protein, filtering based on affinity numbers, crystallizing protein with fragment or fragments-mixture, locating binding sites/binding modes of fragments in 3 dimensional structures, using these spatial-separation & chemical environment of fragments in designing the linkers to join or expand a fragment. Usually IC50 (half maximal inhibitory concentration) of the fragments against a protein target would be weaker – in the order of Milli-moles. Choosing a suitable biophysical technique for screening and filtering the fragments is necessary. Once a few initial compounds or a set of compounds has been generated from the dataset of fragments used for screening, the next steps would follow in the realm of SBDD (Structure Based Drug Discovery) for further expansion of SAR (Structure-Activity Relationship).

At Jubilant Biosys, currently, SPR (Surface Plasmon Resonance) platform is routinely used for fragment screening and the binding mode of the binding fragment is determined by X-Ray Crystallography.
Advantages of FBDD:
Jubilant Biosys provides a wide variety of specialized support for drug discovery services.
If you are looking for quality solutions incorporated with the best practices in the drug discovery domain, feel free to contact us.